Interview de Philippe Roche, réalisée par la Cellule Com’, Juin 2024
L’ARTICLE
Peux-tu résumer cet article en quelques mots ?
Le développement de molécules bioactives ou de médicaments débute généralement par le criblage de fragments pour identifier des hits primaires. Cependant, ces premiers composés doivent être optimisés afin d’améliorer leur affinité et leur spécificité. Cette phase d’optimisation implique plusieurs cycles de modifications chimiques lors desquels des extensions sont ajoutées sur la molécule initiale.
ChemoDOTS est un serveur qui permet de générer l’ensemble des composés facilement synthétisables (en une ou deux étapes) à partir d’un fragment initial, en prenant en compte des règles de chimie médicinale couramment utilisées dans l’industrie pharmaceutique. Le serveur est très simple d’utilisation. Typiquement, les utilisateurs doivent i/ importer le fragment initial ou le dessiner, puis ii/ choisir, parmi les fonctions réactives détectées automatiquement, celle sur laquelle les extensions seront ajoutées. En retour, le serveur propose une liste de réactions chimiques compatibles qui peuvent être sélectionnées pour générer automatiquement l’ensemble des composés synthétisables à partir du fragment initial . Des filtres physico-chimiques peuvent être appliqués pour extraire un sous-ensemble de composés avec des propriétés physico-chimiques spécifiques. Le principe est résumé sur le schéma ci-dessous.
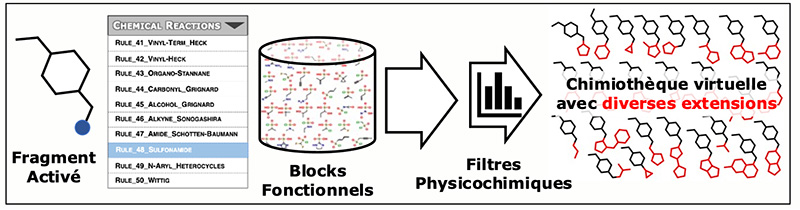
En quoi le serveur ChemoDOTS représente-t-il une avancée significative dans le domaine concerné ?
Ce serveur permet de générer très rapidement un grand nombre de composés qui peuvent être synthétisés facilement et qui représentent une source potentielle de molécules optimisées. Ce type d’approche avait déjà été développé par différentes équipes, dont la nôtre, en utilisant des scripts ‘maison’, mais était réservé jusqu’à présent aux chémoinformaticiens. Ce serveur donne un accès simplifié à la communauté scientifique et ouvre donc la voie à son utilisation par un plus grand nombre. D’ailleurs, depuis qu’il a été mis en ligne, les retours sur ce serveur sont très positifs.
Quels sont les défis qui ont été rencontrés et comment ont-ils été relevés ?
Nous utilisons ce type d’approche dans l’équipe depuis plusieurs années. Un grand nombre de développements avaient été réalisés par Laurent Hoffer, un excellent postdoc qui est resté six ans dans notre équipe. Mais avec Xavier, nous avions l’idée de développer un serveur plus facile d’utilisation depuis plusieurs années. Une première version avait été développée rapidement par Samuel Granjeaud.
Plus récemment, grâce à l’arrivée d’un nouveau thésard, Guillaume Charifi, très performant, notamment dans l’optimisation de code informatique, nous avons pu développer une nouvelle version très conviviale et rapide. Des dizaines de milliers de molécules peuvent être générées en quelques secondes. L’une des difficultés majeures au cours de ces années a été de trouver le bon étudiant pour finaliser ce projet, car le développement d’un serveur web n’est pas forcément considéré comme un projet de recherche en soi, même s’il y a une forte demande de la communauté scientifique.
Quels développements découlant de cette recherche sont envisageables dans un futur proche ?
Nous utilisons déjà ce serveur (ou ses versions en ligne de commande) sur la plupart des projets de l’équipe dans les programmes d’optimisation hit-to-lead. L’organisation de notre équipe, avec les chimistes intégrés aux projets de recherche, se prête tout particulièrement à ce type d’approche. II est actuellement possible d’améliorer un hit initial en seulement quelques semaines. De nouvelles fonctionnalités sont déjà en cours de développement (ajout de nouvelles réactions, d’autres filtres, la possibilité de partir de molécules non fonctionnalisées …).
Nous appliquons également des approche similaires pour la conception de sondes covalentes, qui sont redevenues populaires ces dernières années avec l’approbation de plusieurs médicaments, notamment en cancérologie.
Y a-t-il des analogies simples qui pourraient aider les non-scientifiques à saisir la portée de ce travail ?
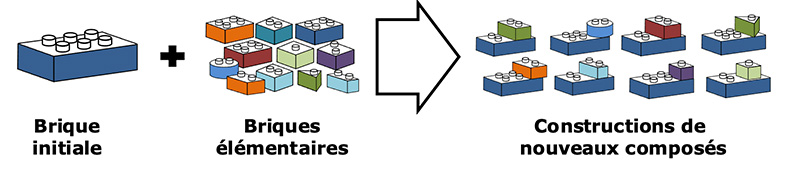
On peut utiliser l’analogie d’un jeu de Lego. Les médicaments (ou n’importe quelle molécule) peuvent être considérés comme l’assemblage de plusieurs briques élémentaires. L’approche ChemoDOTS permet de construire un grand nombre de nouveaux composés en ajoutant des briques élémentaires à une brique initiale, dans le but d’obtenir des molécules plus performantes, comme résumé sur le schéma ci-dessous.
PORTRAIT DE CHERCHEUR
Un peu plus sur toi : ton parcours, ce qui t’a poussé vers la science.
Ma carrière scientifique est loin d’être linéaire, mais elle a été jalonnée de très belles rencontres qui ont souvent influencé mes changements thématiques ou géographiques. J’ai débuté mes études universitaires à Orléans, puis à Toulouse, où je me prédestinais plutôt à la chimie organique. Une rencontre décisive avec le Pr Jean René Pougny m’a conduit à poursuivre un DEA en microbiologie, cependant, j’avais réussi à faire mes deux stages pratiques en chimie de synthèse. J’ai ensuite eu la chance d’arriver au bon endroit au bon moment pour ma thèse sur la symbiose plante-bactérie entre légumineuses et Rhizobium. Durant ces années, j’ai eu une très forte complicité avec Patrice Lerouge, chercheur CNRS. Ce travail a conduit à la caractérisation des premiers facteurs de nodulation et à une période très prolifique qui m’a permis d’obtenir un poste au CNRS en « Biologie Végétale ».
J’ai poursuivi avec un post-doctorat au « Complex Carbohydrate Research Center » aux Etats-Unis. A mon retour en France, souhaitant me consacrer à la caractérisation des récepteurs des facteurs de nodulation, j’ai pris un nouveau virage dans ma carrière en me formant à la modélisation et dynamique moléculaire, notamment dans les laboratoires de David Perahia à Orsay et David Stuart à Oxford. Au fil des années, je me suis définitivement éloigné des facteurs de nodulation pour m’orienter vers la modélisation des interactions protéine-protéine et protéine-ligand. J’ai ensuite intégré l’équipe de Françoise Guerlesquin à Marseille, avant de rejoindre le CRCM en 2012 dans l’équipe de Xavier Morelli, où je suis toujours aujourd’hui.
Je prends beaucoup de plaisir à être dans l’équipe iSCB, car nous partageons de nombreuses valeurs avec Xavier. Je suis d’ailleurs convaincu qu’il adhèrerait à ma devise, qui pourrait être celle de l’équipe iSCB : « Seul on va plus vite, mais ensemble on va plus loin ».
Un peu plus sur toi (bis) : tes activités extra-professionnelles, notamment musicales.
Bien que je ne sois pas issu d’une famille de musiciens, j’ai baigné dans l’univers musical très tôt. J’ai étudié le trombone au conservatoire d’Orléans, mais j’étais surtout intéressé par jouer dans des orchestres. Là aussi, j’ai eu la chance de côtoyer de très bons musiciens au sein de différents ensembles. J’aime l’éclectisme du trombone qui m’a permis de jouer dans des styles musicaux très variés : classique, musique de rue, bandas, orchestres Dixieland, big band de jazz, groupes de salsa ou de rhythm-and-blues. Évidemment, Il m’a suivi lors de mes deux postdocs. Malheureusement, j’ai dû faire une pause pendant plus d’une quinzaine d’années suite à de forts acouphènes.
Récemment, j’ai renoué en jouant avec le Big Band du CNRS, l’orchestre symphonique de l’université (OSAMU) et quelques formations locales. Actuellement, mon activité musicale est à nouveau au point mort mais je ressens toujours un manque quand j’assiste à des concerts … alors qui sait.
En savoir plus sur l’article